Quickstart guide¶
This guide will demonstrate how to use immuneSIM and how to generate a simple simulated immune repertoire.
Overview¶
The goal of the immuneSIM simulation is to in silico generate human and mouse B- and T-cell repertoires with user-defined properties to provide the user with custom native or aberrant immune receptor sequence repertoires to benchmark their repertoire analysis tools. The simulation algorithm implements an in silico VDJ recombination process with on-the-go annotation of the generated sequences and if enabled by the user somatic hypermutation (SHM) and motif implantation. With a wide range of user-modifiable parameters, a uniquely diverse set of repertoires can be created. The parameters include: Clone count distribution, Germline Gene Usage, Insertion and Deletion Occurrence, SHM likelihood and Motif Implantation.
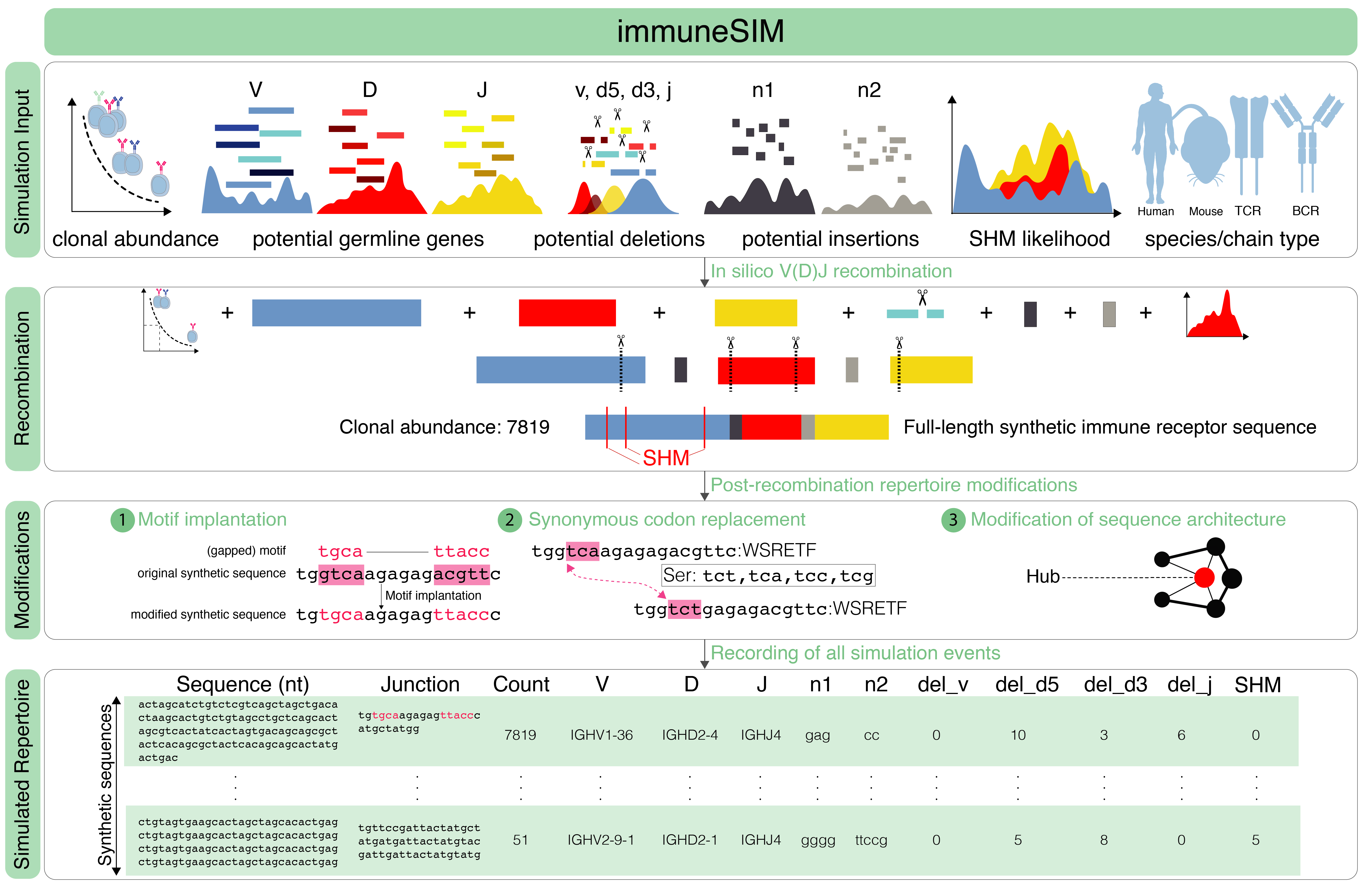
The ImmuneSIM in silico VDJ recombination is based on our understanding of the VDJ recombination process and includes frequency-based selection of a V, D and J genes and insertions and deletion events in the VD and DJ junctions.
Prerequisites¶
To be able to run the code, the following prerequisites are:
- R >= 3.4.0.
- Imports: poweRlaw, stringdist, Biostrings, igraph, stringr, data.table, plyr, reshape2, ggplot2, grid, ggthemes, RColorBrewer, Metrics, repmis
Installing immuneSIM¶
The package can be installed in R (via CRAN or GitHub):
- Check if all the prerequisites are fulfilled/installed.
- Execute the following lines in R:
#install the devtools package
install.packages("immuneSIM")
Alternatively, installation via GitHub is possible in R:
#install the devtools package
install.packages("devtools")
#load devtools and install immuneSIM from github
library(devtools)
install_github("GreiffLab/immuneSIM")
Workflow of the quickstart simulation¶
The quickstart simulation using ‘immuneSIM’ generates a repertoire of a chosen size for a given species and receptor combination. It does not include somatic hypermutation and motif implantation.
The repertoires are simulated by in-silico VDJ recombination. Each repertoire will consist of a user-predefined number of fully annotated immune receptor sequences.
The user can generate pdfs summarizing the major features of the generated repertoire that includes: VDJ usage, positional amino acid frequency and gapped-k-mer occurrence.
Performing the analysis¶
In the quickstart.R, we provide a simple example of murine B-cell repertoire generation based on standard (experimental) parameters:
library(immuneSIM)
sim_repertoire <- immuneSIM(
number_of_seqs = 1000,
species = "mm",
receptor = "ig",
chain = "h",
verbose= TRUE)
save(sim_repertoire,file="sim_repertoire")
#output plots on repertoire (Note: you need to specify output directory)
plot_report_repertoire(sim_repertoire, output_dir="my_directory/")
Output plotting function¶
The above example ends with the plot_report_repertoire function which outputs pdfs of the length distribution, amino acid frequency of most abundant length and VDJ usage plots.
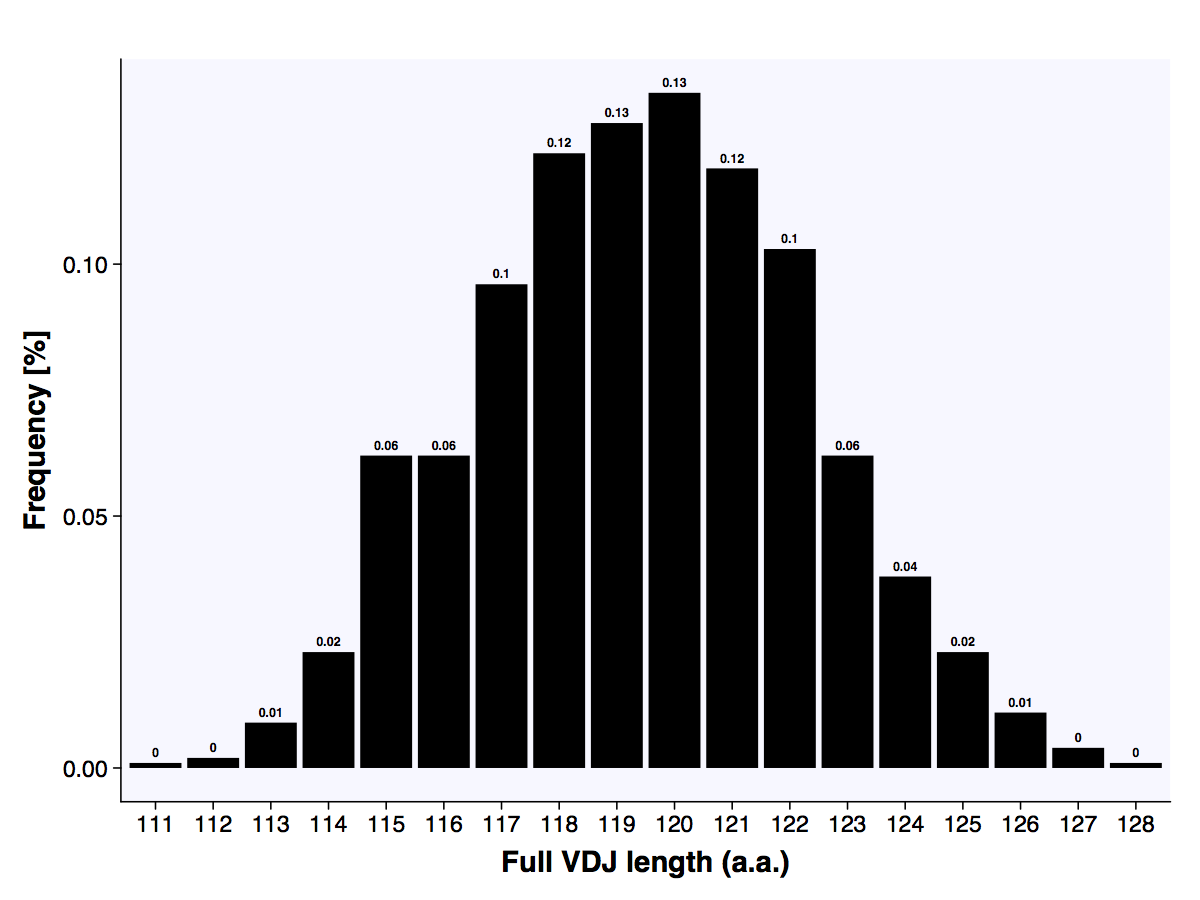
The length distribution of the full VDJ sequences (a.a).
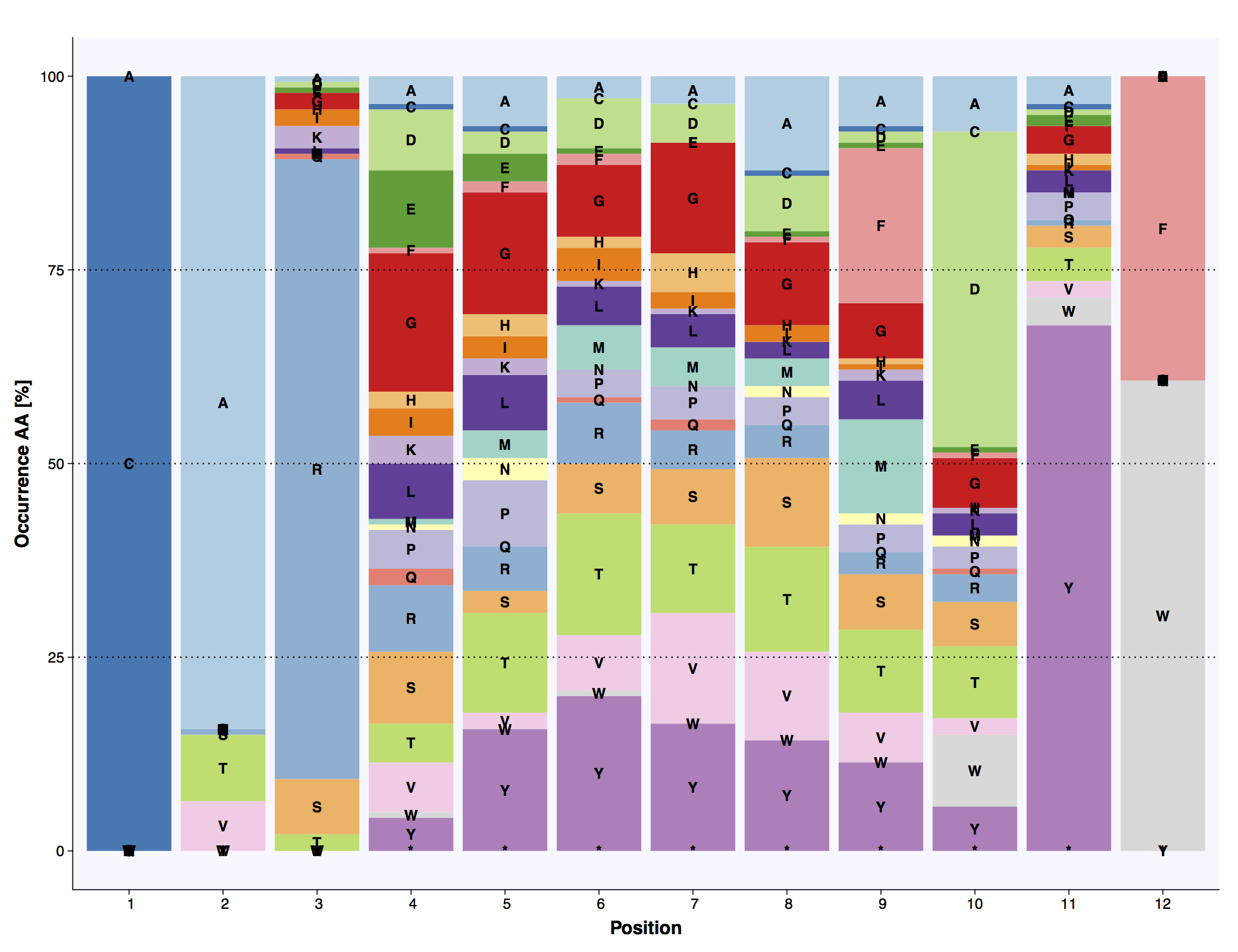
The positional amino acid frequencies of the most abundant CDR3 length in the simulated dataset.
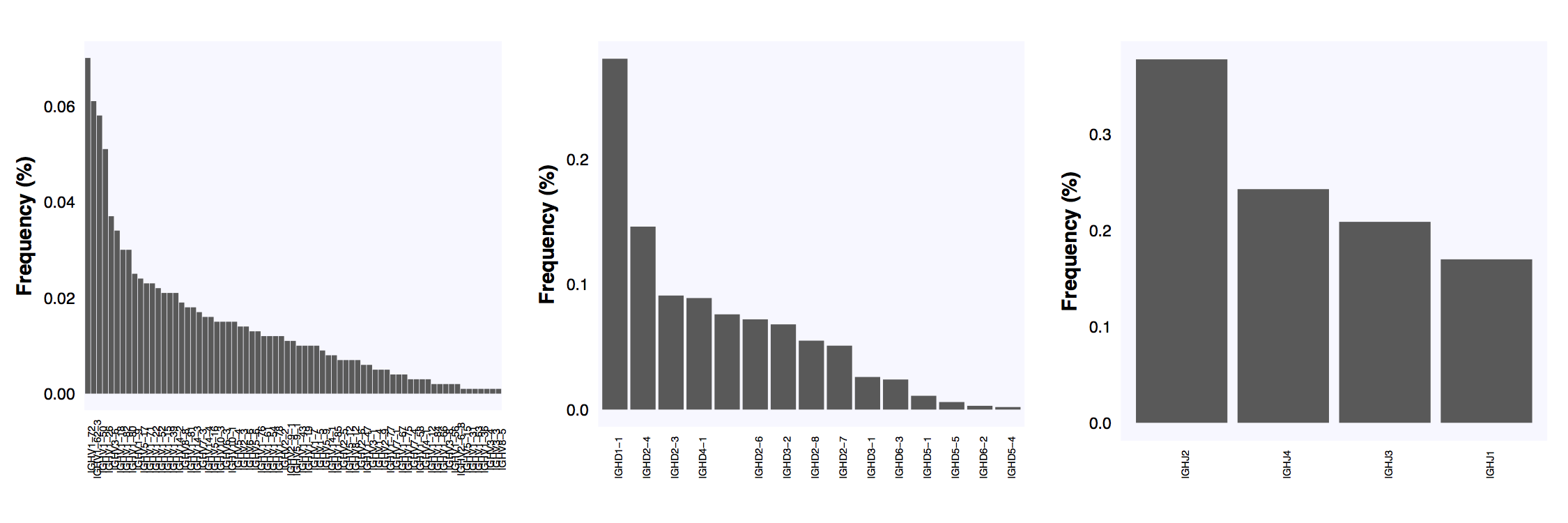
The V, D, J usage in the simulated repertoire.