Simulation¶
The workflow of a standard immuneSIM simulation¶
The analysis will consist of the following steps:
- Simulation of the immune repertoire (including SHM)
- Post simulation modifications
- Output of report comparing repertoire to naive experimental repertoire.
The repertoires are simulated by in-silico VDJ recombination. Each repertoire will consist of a user-predefined number of fully annotated immune receptor sequences. During the simulation process SHM can be performed based on the previously published AbSIM package [1]. Following the simulation process there are several options to introduce additional biases in the repertoire through Post simulation modifications. Finally the user can generate a report about the generated repertoire that includes: VDJ usage, positional amino acid frequency and gapped-k-mer occurence (See: Report generation). The entire process is summarized in a flowchart below (See: Flowchart immuneSIM)
library(immuneSIM)
number_of_sequences <- 1000
mm_igh_sim <- immuneSIM(number_of_seqs = number_of_sequences,
vdj_list = list_germline_genes_allele_01,
species = "mm",
receptor = "ig",
chain = "h",
insertions_and_deletion_lengths = insertions_and_deletion_lengths_df,
user_defined_alpha = 2,
name_repertoire = "mm_igh_sim",
length_distribution_rand = length_dist_simulation,
random = FALSE,
shm.mode = 'none',
shm.prob = 15/350,
vdj_noise = 0,
vdj_dropout = c(V=0,D=0,J=0),
ins_del_dropout = "",
equal_cc = FALSE,
freq_update_time = round(0.5*number_of_sequences),
max_cdr3_length = 100,
min_cdr3_length = 6,
verbose = TRUE,
airr_compliant = TRUE)
save(mm_igh_sim,file="mm_igh_sim")
in-silico VDJ recombination¶
To enable a simulation in which the prediction of the immune status can be performed for synthetic data, immuneSIM introduces an in-silico VDJ recombination algorithm.
VDJ Recombination includes
- sampling clone count (Parameter 4: Clone count distribution)
- sampling V, D and J genes (Parameter 5: V,D,J germline gene frequencies)
- sampling insertions and deletions (Parameter 6: Insertion and deletions)
- sampling SHM profile (Parameter 7: Somatic hypermutation likelihood)
- introducing signals (Parameter 10: Motif implantation)
Output format¶
The immuneSIM function outputs an R dataframe containing 20 columns and rows equal to the number of sequences simulated. Per sequence immuneSIM provides the following information: * Full VDJ sequence (nucleotide and amino acid): sequence, sequence_aa * CDR3 junctional sequence (nt and aa): junction, junction_aa * VDJ genes used in the recombination event: v_call, d_call, j_call * Nucleotide insertions VD and DJ: np1, np2 * Length of deletion in V, D and J genes: del_v, del_d_5, del_d_3, del_j * CDR3 subsequences from V,D and J genes: v_sequence_alignment, d_sequence_alignment, j_sequence_alignment * Clonal frequency/count information: freqs, counts * Summary of SHM event simulated: shm_events * Given name of repertoire: name_repertoire
VDJ pool¶
The in-silico VDJ recombination process draws from a pool of germline V, D and J genes. Each immune receptor sequence simulation event starts by sampling a V, D and J germline gene sequence based on its assigned frequency, which is either based on experimental data or the user’s preference. To facilitate ease of use, the immuneSIM package includes a library of germline gene datasets for mouse and human BCR and TCR germline genes for both heavy and light and beta, alpha chain (based on IMGT database). Apart from the sequence and annotation information, these datasets include frequencies for each gene as observed in experimental datasets [2].
However, the user is free to add additional datasets with different frequency distributions or with data for species not included so far. (See: Introducing new germline gene frequencies)
Insertions and deletions¶
In the recombination process additional diversity is created through nucleotide insertions and deletions. Deletions occur at the 3’ end of the V gene, on 5’ and 3’ end of the D gene as well as on the 5’ end of the J gene. Nucleotides are inserted at the junction of the V and D gene and also between the D and J gene. ImmuneSIM randomly samples a vector of deletion lengths c(del_V,del_D5,del_D3,del_J), subsequently it samples insertions of lengths complementing the resulting V,D and J sequences such that an in-frame sequence results. (See: Parameter 6: Insertion and deletions).
NOTE: Due to package-size limitations on CRAN, the dataframe insertion_and_deletion_lengths_df contained in the immuneSIM package is only a subset of 500’000 entries of a larger dataset
with 11’363’603 entries. The full dataet is provided on GitHub and can be retrieved using the load_insdel_data() function.
Clone count¶
The clone count is simulated to result in a power law distribution of counts, the user can set the alpha parameter that controls the evenness and also has the choice to create a clone count that is equal across all clones. [3] (See: Parameter 4: Clone count distribution)
Somatic hypermutations¶
The Somatic hypermutations are simulated based on the previously published AbSIM R package [1]. (See: Somatic hypermutation)
Post simulation modifications¶
In this step a variety additional biases can be introduced. This includes:
- Introducing (antigen-specificity simulating) signals. (See: Motif implantation)
- Introducing a codon bias. (See: Parameter 12: Codon bias)
- Modifying the sequence similarity structure. (See: Parameter 11: Sequence similarity)
Report generation¶
immuneSIM provides the user with a means to output figures in pdf format summarizing the generated repertoire, either by itself or in comparison to a reference repertoire. (See: Report generation)
Flowchart immuneSIM¶
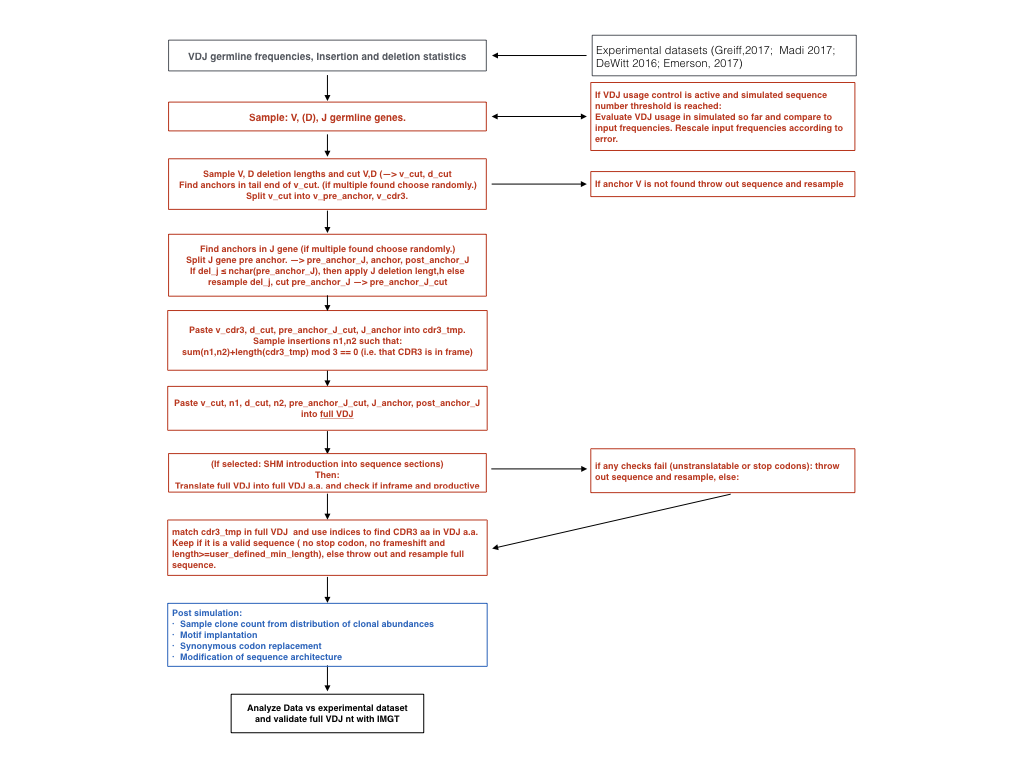
The step by step breakdown of the in-silico VDJ recombination as performed by immuneSIM.
| [1] | (1, 2) Comparison of methods for phylogenetic B-cell lineage inference using time-resolved antibody repertoire simulations (AbSim), Yermanos et al., Bioinformatics, 33(24), 2017, https://academic.oup.com/bioinformatics/article/33/24/3938/4100159 |
| [2] | Systems Analysis Reveals High Genetic and Antigen-Driven Predetermination of Antibody Repertoires throughout B Cell Development, Greiff et al., Cell Reports, 19(7), 2017, https://www.sciencedirect.com/science/article/pii/S221112471730565X |
| [3] | Fitting Heavy Tailed Distributions: The poweRlaw Package, Gillespie, Journal of Statistical Software, 64(2), 2015, https://www.jstatsoft.org/article/view/v064i02 |